

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
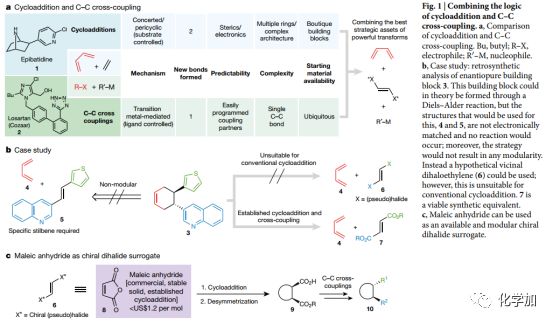
Fig.1将环加成和碳碳键偶联结合a.对比环加成和偶联b.逆合成分析c.马来酸酐作为手性二卤替代品
与一次形成一个键(多为sp2碳)的偶联反应相比,一次形成多个碳碳键的环加成反应有着天然的优势(Fig.1a)。但是,偶联反应的优势在于比环加成反应更具多样性及模块化。生物碱地棘蛙素(1)和市售降压药物科素亚(2)的合成就足以阐明这两种反应的特点。由于地棘蛙素有效的镇痛效果,化学家们已经对其进行了六十多次全合成,其中有31次使用了环加成作为关键步骤构建其吡咯烷核心骨架,并经过多步转化后引入吡啶环。科素亚畅销于全球,骨架结构中不含任何立体中心和拓扑结构,其发明和最终的工业化生产都使用了碳碳键偶联反应。偶联反应的汇聚式组装和模块化构建使得多种类似物得以简单合成。
化合物3的结构往往让人想起狄尔斯-阿尔德反应(Fig. 1b),但由于片段4和5的电性不匹配,反应并不能如愿发生。但即使可以发生,此类反应也缺乏模块化构建的特点。为了解决该问题,作者设计了可代替5的延胡索酸酯类型亲烯体(7),将狄尔斯-阿尔德反应和自由基偶联反应(RCC)结合起来,实现多个立体中心及模块化构建。
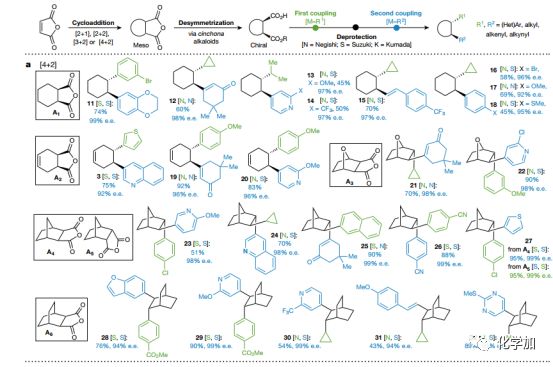

Fig.2 结合环加成和碳碳键杂交偶联的底物适用范围
选择商业可得的廉价化合物马来酸酐代替带有手性辅基化合物6参与多种环加成反应([2+1]、[2+2]、[3+2]、[4+2]),然后用手性碱催化去不对称醇解,得到具有连续手性中心的环加成产物。然后自由基偶联、水解、二次自由基偶联后便得到目标化合物。由于自由基偶联反应底物的广泛性,可以实现多种结构的构建,作者在文中展示了包括天然产物以及工业化生产实例在内的80多个例子。
首先从狄尔斯-阿尔德反应开始(Fig. 2a)。狄尔斯-阿尔德反应构建的A1-A6在邓组开发的去不对称条件下(以奎宁或奎尼丁为路易斯碱)催化不对称醇解,得到高对应选择性的单酸单酯底物,而后连续进行Negishi和Suzuki类型的RCC反应。通过此过程,可将简单廉价的狄尔斯-阿尔德反应产物A1转化为具有高对应选择性以及高附加值的产品(11到63,涵盖了芳香基、杂芳香基、烯烃、具有张力的环丙基、环丁基以及简单片段甲基和异丙基)。大多情况下狄尔斯-阿尔德反应往往需要缺电的烯烃和富电的双烯,使得反应的多样性大大受限,但文中的方法可以构建常规狄尔斯-阿尔德反应不能得到的产物(不论消旋还是手性)。此外,在传统狄尔斯-阿尔德反应中,当双烯片段里有多个烯烃存在时,无法有效地控制反应。但作者通过模块化构建解决了此类问题(12、15、19、21、25 和31)。有趣的是外型和内型异构体A4、A5,在调整反应次序后均可得到相同的产物27。
此方法可应用于多种环加成反应中,如[3+2]、[2+2]和[2+1](Fig. 2b-d),且偶联片段与前文所述的狄尔斯-阿尔德反应砌块一样多种多样。比如含吡咯烷的体系更可直接应用到药物合成中,大约有四十多种药物含有此结构。通过Pd催化可以较为容易地得到的环戊烯B3结构,很难通过其他方法合成。通过[2+2]环加成构建的C1 和C2,其对电子要求严苛,难以满足产物多样化的需求,且光催化生成的1,2-二取代环丁烷结构会产生难以分离的消旋化物质(83-86,源于C1)。有极高的应用价值的四取代手性环丁烷,多个二聚和伪二聚环丁烷天然产物含有此类结构,比如87。从D1和D2 骨架开始的手性环丙烷类化合物构建也有极高的难度。这些难以通过传统手段获得的多种环结构,均可以通过文中开发的方法获得。
作者还使用此方法合成了六个天然产物和药物前期或后期重要中间体(Fig. 3a-f)。
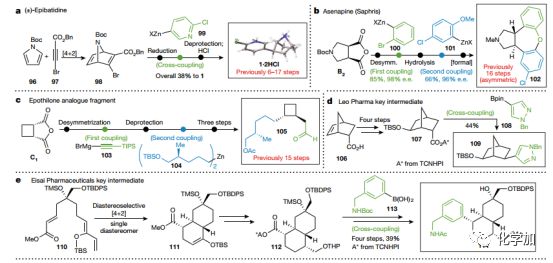
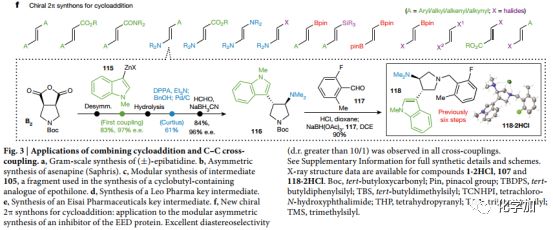
Fig.3 结合环加成和碳碳键杂交偶联反应的应用a.克级合成(±)-epibatidine. b.不对称合成asenapine (Saphris). c.模块化合成中间体 105d.合成Leo Pharma的关键中间体 e.合成Eisai Pharmaceuticals的关键中间体f.新手性2π 合成子应用于环加成
比如前文所说的生物碱1,只需要5步便可得到,关键步偶联反应产率极高,且总产率高达36%(Fig. 3a)。
被FDA列为抗精神病药物的Saphris (阿塞那平,102;Fig. 3b)是一个苯环上氯原子导致的手性化合物,目前其最短的不对称合成路线为16步,而此方法可以将其不对称合成路线简化为6步,且两个偶联关键步的产率分别是85%和66%。
Epothilone是一种具有细胞微管稳定活性的天然产物,为FDA批准抗肿瘤药物Ixabepilone的重要中间体,因此是合成研究热点。中间体105(Fig. 3c)从报道的15步被作者简化到8步合成。
药物化学中降冰片烯被认为是苯基的生物电子等排,由于缺乏有效的模块化构建不同取代基的方式,其应用一直被限制。在和Leo Pharma的合作中作者用此方法合成了一个正在进行项目的重要中间体(109;Fig. 3d)关键步偶联产率44%,且反应中首次使用硼酸酯作为RCC反应的底物,通过酸根型配合物(Ate complex)实现。
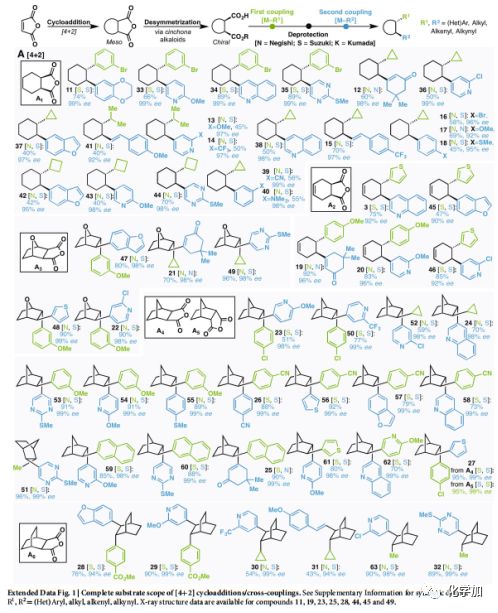
扩展数据Fig.1完整[4+2]环加成/杂交偶联底物范围
Eisai Pharmaceuticals一个正在进行的项目中,中间体114的关键结构的芳基部分通过文中方法引入。用Corey的噁唑硼烷催化剂催化构建十氢化萘结构111,然后在酯基部分引入芳基得到114。
文中开发的方法不仅仅局限于碳碳键的形成,通过与亲核官能团化和脱羧官能团化结合,可以实现多种多样性结构的合成(Fig. 3f)。例如可以通过2π烯胺合成子合成118(以前以消旋[3+2]手段得到,总产率1.9%)。使用文中的方法,以B2为原料,通过偶联反应以及Curtis重排得到单一手性的目标产物,总产率为13%。
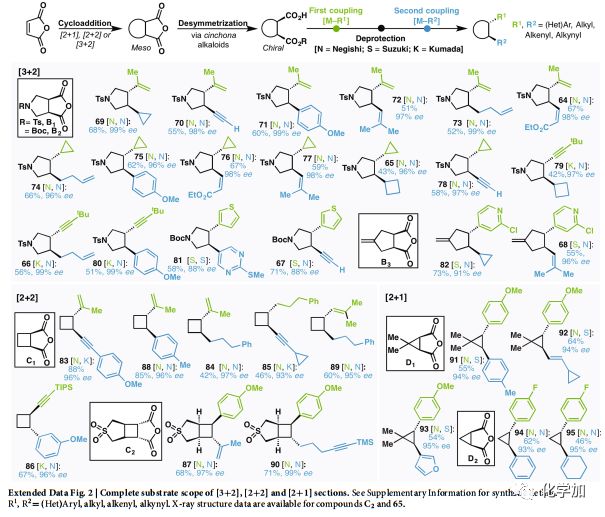
扩展Fig.2完整[3+2],[2+2],[2+1]类型底物范围
小结:正如各种武功都有自己的弱点一样,文中方也存在局限性。首先其局限性分别是环加成和偶联反应各自的局限性。其次顺式-1,2-二取代产物目前无法通过此方法得到,除非通过异构化反应逐个尝试,或许通过配体控制的RCC反应可以解决此问题。本文将经典的环加成反应和新发展的RCC偶联反应结合,可以得到传统手段很难得到的很多结构,对天然产物和药物合成有重大的意义。
原文:Building C(sp3)-rich complexity by combiningcycloaddition and C–C cross-coupling reactionshttps://doi.org/10.1038/s41586-018-0391-9
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
