

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
自1994年以来,化学家们从苔类植物中分离和表征了一组具有并环呋喃酮的非常复杂的二萜化合物,如pallavicinin (1)、pallavicinin (2)和pallambins A-D(3-6)(图1A),它们含有4-6个拥挤环和7-10个连续手性中心,包括1-2个全碳季碳中心,其全合成极具挑战。然而,这类天然物并没有显示出显著的生物活性,可能是因为它们自然来源极少,从而限制了其生物评价。因此,对这些二萜类化合物进行简明的化学合成可以提高这些分子的可用性,以便进行全面的生物评估。
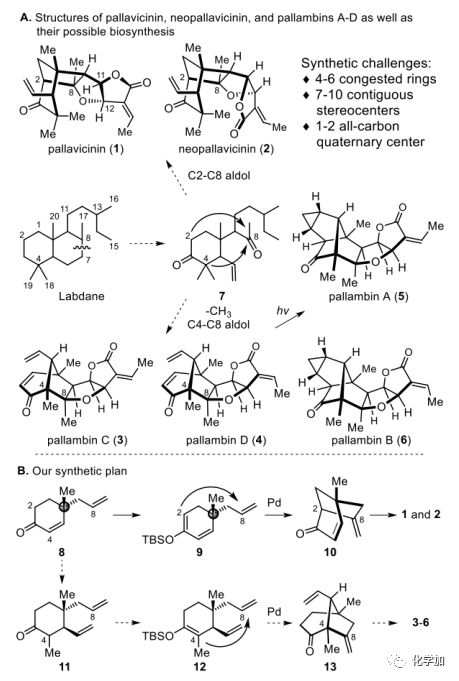
图1. 目标分子的结构、生源合成及合成计划(图片来源:Angew.Chem. Int. Ed.)
Asakawa提出了这些天然产物的合理生物合成途径(图1A)。labdane型二萜类化合物C7-C8键的断裂提供了共同的前体7。1和2是通过分子内Aldol反应重建C2-C8键而产生的。3和4是通过去甲基化和C4-C8键重建形成的。娄红祥等人进一步证明了3和4通过光诱导的双自由基重排到5和6的相互转化。
1-6全新且极具挑战性的分子结构吸引了合成化学家的广泛关注,黄乃正课题组率先从Wieland-Miescher酮合成了(±)-1至(±)-4。Carreira等人在2015年以Diels-Alder反应实现了 (±)-5和(±)-6的首例全合成。同年,贾彦兴课题组从已知的手性环己烯酮8(图1B)出发完成了(-)-1和(+)-2的不对称合成。2016年,Baran课题组报道了一种优雅的(±)-3和(±)-4的合成,且没有使用保护基。
由于作者最近成功地从8合成了(-)-1和(+)-2,并受上述生物合成假说启发,作者设想8可以作为合成1-6的公共中间体。化合物8很容易转化为12,经Pd-催化氧化环化得到[3.2.1]双环酮13,13可转化为3-6(图1B)。因此,可以实现1-6的多样性合成。在此,作者报道了在不使用保护基的情况下,第一次不对称合成pallambins A-D(3-6)。
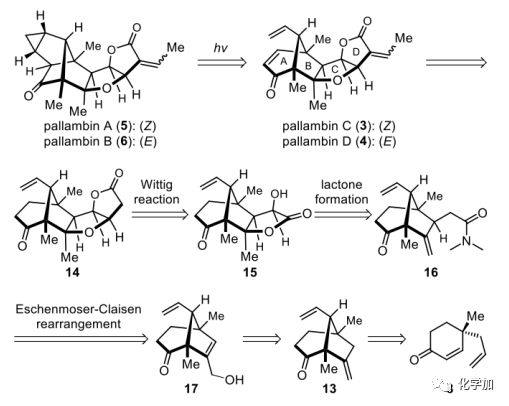
图2. pallambins A-D的逆合成分析(图片来源:Angew.Chem. Int. Ed.)
3-6的逆合成分析如图2所示。作者设想5和6可以通过娄红祥的3和4的光诱导相互转化很容易地得到。反过来,3和4可以从四环化合物14得到,其中BCD三种不同的环系逆推至三种不同的环化方式。因此,D环由内酯15的分子内Wittig反应而形成。C环是由酰胺16形成内酯形成的,酰胺16可以通过烯丙醇17的Claisen重排而生成,烯丙醇17可以从双环化合物13很容易地合成,13可以从8得到。
全合成从已知的手性化合物8开始,8可以按Stoltz的方法以84% ee轻易制备。乙烯基与8共轭加成,然后甲基化,得到酮11,在C5处的非对映体比(dr)为3:1。用Et3N和TBSCl对酮11进行处理,得到相应的热力学TBS烯醇醚,再在优化的Pd-催化氧化条件下,得到了期望的双环[3.2.1]辛烷体系13,产率64%。用m-CPBA对13的富电子双键进行化学选择性环氧化,得到相应的环氧化产物,在1,3-二甲基咪唑啉-2-酮(18)存在下用PTSA处理,得到所需的烯丙醇17。稳健得到17以后,接着集中于C环的构建。在甲苯中115 °C下加热17和N,N-二甲基乙酰胺二甲基缩醛,以单一对映体的形式得到了理想的γ,δ-不饱和酰胺16,产率为88%。在回流乙醇中用H2SO4处理16,得到内酯19,收率84%。
为了在C11处安装羟基,必须保护19中的酮。然而,对分子模型的进一步观察表明,由于空间拥挤的A/B环系屏蔽了C3-酮基的α-位,游离C3-酮基可能不会影响α-羟基化反应。因此,作者大胆地选择直接引入C11羟基,而不保护C3酮。在对几种条件进行测试后,作者发现,用3当量LiHMDS对19去质子化,再加入戴维斯试剂(20),得到a-羟基内酯21,为单一的立体异构体。由于19的高张力笼状结构,羟基化反应只发生在位阻较小的凸面上,与C8甲基成顺式。这与天然产物所需的立体化学是相反的。因此,采用氧化/还原策略对C11羟基的构型进行了翻转。用DMP氧化21得到相应的酮内酯,并用LiAlH(Ot-Bu)3进行了完全的化学和立体选择性还原,得到所需的醇15为单一立体异构体,收率为99%。
得到了三环化合物15,接着引入最后一个环系。在间二甲苯中160 ℃下用(三苯基磷酰亚甲基)烯酮和15反应,完成了a-羟基的酰化反应和随后的分子内Wittig反应,得到了四环酮酯22。然后进行了四环酮酯双键的化学和立体选择性还原。经过广泛的实验,作者发现CuI与Red-Al反应原位生成CuH还原22,得到目标产物14,产率为94%,其结构经X-射线晶体分析确证。

图3. pallambins A-D的全合成(图片来源:Angew.Chem. Int. Ed.)
在这个阶段,作者需要安装C1,C2双键;然而,这是非常具有挑战性的。首先研究了酮直接脱氢的一步法,但是,用IBX氧化酮14,以及Pd-催化脱氢等其他方法,都没有得到24。最后,14与Py·HBr3在HOAc中溴化得到单一立体异构体23,产率为80%。用Li2CO3/LiBr、DBU或其它碱对23的脱溴化氢反应进行了筛选。反应在低温(<80 ℃)下不发生,在高温(>80 ℃)下原料分解,未观察到预期产物24。这些结果表明,23和(或)24可能对高温条件下的碱敏感。因此,需要开发一种温和的脱溴化氢方法。
在Pd-催化Heck反应温和的反应条件和反应机理的启发下,作者认为a-溴酮23对Pd(0)物种的氧化加成可以得到相应的烷基钯物种,经过β-氢消除能得到所需的烯酮24。经过广泛的试验,作者发现在优化的条件下(DMSO中的Pd(OAc)2、PPh3和Et3N)处理23,可获得所需产物24,收率为41%,Heck型产物25的收率为38%。在24上装上亚乙基,以30%的产率得到目标分子pallambin C(3),51%的产率得到pallambin D(4),合成产物3和4的物理数据与文献报道一致。
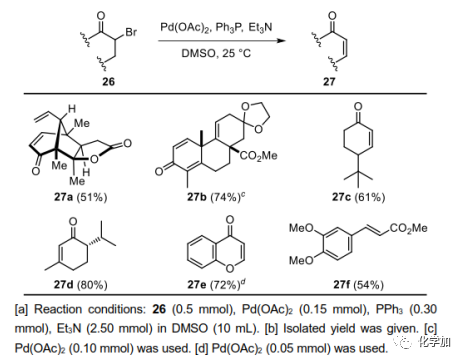
图4. a-溴代酮脱溴化氢(图片来源:Angew.Chem. Int. Ed.)
最后,根据娄红祥课题组的研究,在紫外光下将pallambin C(3)和D(4)转化为pallambin A(5)和B(6)。有趣的是,在较温和的条件下(紫外灯,8 W),在25 ℃的CH2Cl2中,3和4的单独辐照分别产生5和6,而在5和6之间以及3和4之间没有双键顺式/反式异构化。这一结果清楚地表明,在∆13(14)处,双自由基重排所需的能量比顺式/反式异构化所需的能量要低。合成产物5和6的物理数据与文献报道一致。
总结:北京大学贾彦兴课题组在不使用保护基的情况下,从已知的环己烯酮8出发,用15-16步反应完成了pallambins A-D的首次不对称全合成。这种无保护基合成的成功主要取决于几个高度的化学和立体选择性反应:Pd-催化氧化环化构建[3.2.1]-双环结构,Claisen重排/内酯形成序列构建C环,分子内Wittig反应形成D环。在此过程中,作者还开发了一种温和的a-溴代酮脱溴化氢方法。
撰稿人:诗路化语
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
