

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家

TOC图(来源:Angew. Chem. Int. Ed.)
酶是自然进化形成的高效生物催化剂,一般适用于天然底物和相应的生化反应。随着可持续合成化学的理念深入推进,有机化学家们开始探索天然酶的非天然反应性,即“催化杂泛性”。近年来,这方面的研究已经取得了显著的进展,尤其是铁辅因子依赖的P450酶家族,结合定向进化等生物技术,其催化活性已经从天然的氧化反应拓展至不对称卡宾/氮宾转移等合成反应(2018年诺贝尔化学奖)。相比之下,铜氧化酶作为一大类氧化还原酶家族,在不对称催化领域的应用还十分少见。考虑到小分子铜配合物在化学催化方面的多样性功能,研究铜氧化酶的催化杂泛性以实现非天然不对称催化反应是一个有意义的课题。
近年来,氧化交叉偶联反应已经成为绿色高效的合成工具,可以将两个富电子亲核试剂通过成键连接,避免了底物预功能化,具有显著的原子经济性。不对称氧化交叉偶联的难点在于同时控制氧化步骤的化学选择性和偶联步骤的对映选择性(图1a)。已知的文献方法主要是基于化学氧化或光化学/电化学氧化与手性过渡金属或有机催化剂的组合。氧化还原酶具有催化不对称氧化偶联的潜力,如自然界中铜依赖漆酶催化的γ-萘酮同分异构体(M)-ustilaginoidin A的不对称同源二聚反应,具有专一的区域和对映选择性(图1b)。然而,非天然底物的酶促对映选择性氧化偶联反应仍然很少。去年,Narayan小组利用工程化P450酶实现了酚类分子生物催化交叉偶联反应,为轴手性联芳基分子的合成提供了有效策略(图1c)。
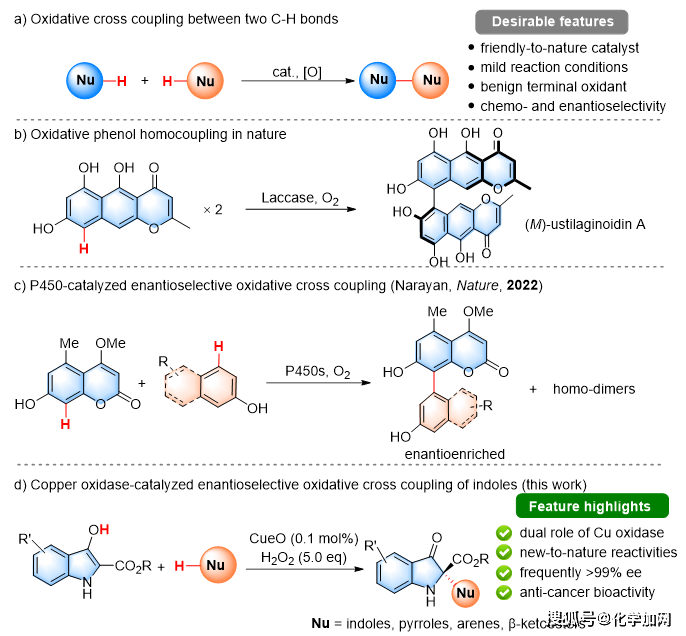
图1. 生物催化氧化交叉偶联反应(来源:Angew. Chem. Int. Ed.)
多铜氧化酶(MCOs)广泛存在与自然界中,一般具有较低的底物特异性,可接纳多种天然底物,其中主要类别是酚类化合物。MCOs通过单电子转移机制催化酚的氧化反应,生成易二聚或寡聚的活性苯氧自由基,难以控制氧化反应下游的反应过程。这种性质导致漆酶催化的酚类氧化偶联反应中常形成多种区域异构体和立体异构体的混合物,立体化学的调控往往依赖于额外的辅助蛋白。
钟芳锐团队致力于绿色仿生和生物催化领域的研究,利用化学原理驱动酶催化选择性合成的理念,发展了含有非天然光敏活性基团的人工光敏金属酶(J. Am. Chem. Soc. 2021, 143, 617)和“三重态光酶”(Nature, 2022, 611, 715)。基于铜催化氧化偶联反应的化学机制,发展了多铜氧化酶催化的多酚氧化环加成反应(Green Chem, 2022, 24, 5598.)
在之前的酶催化工作基础上,构思在酚类底物中引入额外的氢键供体/受体,可能与酶的手性腔室形成有益的相互作用,从而实现对映体诱导的可能性。由此发展了对多铜氧化酶CueO催化两种不同类型吲哚之间的不对称氧化偶联反应(图1d)。这种方法具有良好的底物通用性、优异的化学选择性/对映选择性和温和的反应条件。
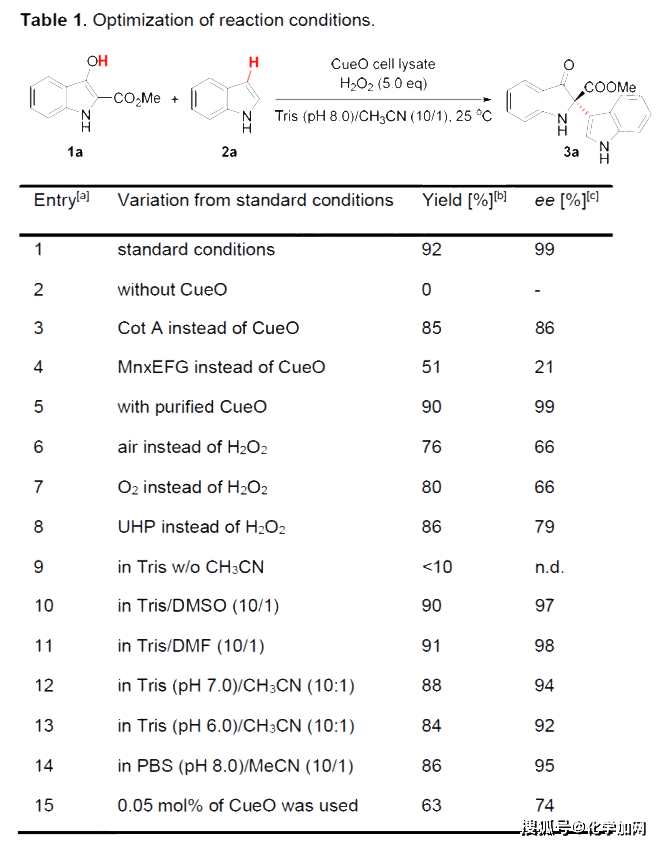
作者以3-羟基吲哚-2-羧酸1a和吲哚2a作为偶联反应物进行条件优化(表1)。由于后者带有的缺电子酯基会显著降低氧化反应的活性,这有利于氧化步骤的化学选择性,而偶联产物3a含有的2,2-二取代吲哚-3-酮骨架也常见于一些生物活性分子。当铜酶CueO进行催化反应时,模型反应在空气中顺利进行,并且在适当的反应条件下(0.1 mol% CueO,5.0当量双氧水,25℃,24小时)可以得到产物3a,收率为92%,对映选择性为99%。其他多铜氧化酶,例如Cot A和MnxEFG,也能催化该反应,但结果较差。令人高兴的是,CueO细胞裂解液的结果与纯化酶的结果几乎相同,因此大大减少了繁琐的蛋白质纯化过程,尽管铜氧化酶在天然生物系统中通常使用分子氧作为最终电子受体,当前体系中过氧化氢的效果优于分子氧和尿素过氧化氢,这可能是由于双氧水与Cu2+离子的配位作用带来了更合适的蛋白质构象。有机共溶剂的使用解决了底物的溶解度,但需要保持弱碱性以避免酸促进背景偶联反应。例如,当pH≤7.5时,酸催化背景反应就会显著提高,导致ee值降低。
表1. 反应条件优化 (来源:Angew. Chem. Int. Ed.)
该CueO催化不对称交叉偶联具有较好的底物兼容性(图2)。苯环上具有不同电子和立体性质的取代基的吲哚酚、各种含卤素、CF3或CN的吲哚亲核试剂、富电子类吡咯、1,3,5-三甲氧基苯和1,3-二甲氧基苯也同样适用。除此之外,亲核试剂的类型还可以进一步扩展到sp3杂化碳亲核试剂,如环状和非环状β-酮酸酯,相应的产物均具有极好的立体选择性。部分底物的放大实验(0.3-1.0 mmol)表明产物的产率和光学纯度可以基本保持不变(产物3a、3d、3f和4c)。
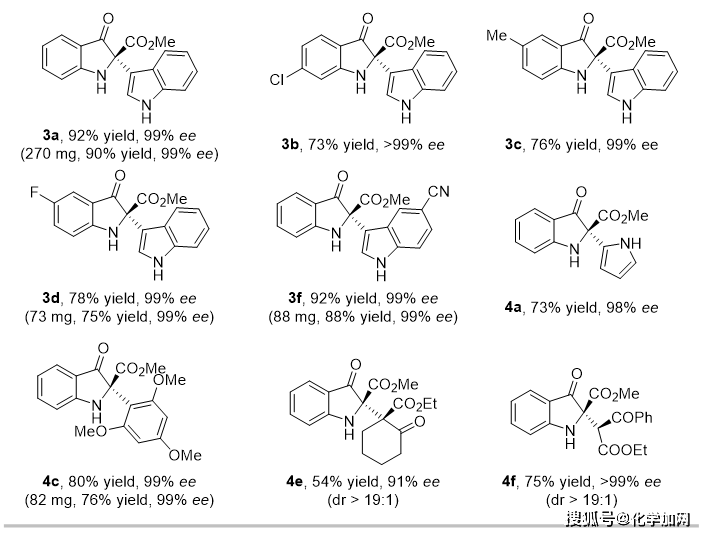
图2. 代表性底物的结果(图片来源:基于原图重新编辑)
多铜氧化酶催化氧化酚类底物一般通过自由基机理进行。为了对该反应的机理进行更深入的研究,作者在模型反应中添加了自由基清除剂如叔丁基羟基甲苯(BHT)。结果显示,产品3a的生成几乎被完全抑制,高分辨质谱(HRMS)检测到化合物4的生成(图3a)。这一结果表明,反应可能经历了酚氧自由基中间体的形成。另一方面,当不加2a时,高分辨质谱检测到亚胺中间体5的生成。不过,该中间体非常不稳定性,无法分离。相比之下,吲哚2a单独无法被CueO氧化(图 3b)。通过两个底物的氧化还原电势可以推测,CueO首先催化氧化吲哚酚1a形成中间体5,随后与吲哚2a发生亲核加成。由于相关文献表明,加成步骤需要酸活化,作者推测CueO应该起到了催化氧化和酸催化的双重角色。CueO的蛋白晶体结构(PDB:3OD3)包含四个铜原子:一个类型1铜(T1 Cu)负责底物氧化,一个三核簇负责氧还原。T1 Cu与两个组氨酸、一个半胱氨酸和一个轴向配体甲硫氨酸形成三角配位模式,这不利于它提供额外的路易斯酸配位。而从底物谱的结果中可推测,平面型中间体5具有高度的对映面选择性。由此,作者通过对CueO晶体结构的活性位点进行仔细分析,将中间体5作为底物进行对接。模拟表明,中间体5紧密结合到结构域2界面。利用LigPlot + v.2.2.7可视化分析表明(图3c),中间体5位于一个由Ala153、Asp187、Lys188、Lys189、Asn240等残基围成的口袋。其中最显著的弱相互作用来自于5的酯基与Asn240的酰胺N-H和Lys188的质子化氨基间(图3d),这些相互作用可能对交叉偶联反应的立体化学控制至关重要。为了验证这一点,这两个残基被突变成丙氨酸,两个突变体CueO_K188A和Cueo_N240A的选择性显著变差(分别为86% ee和55% ee),产率也有所降低,这佐证了上述假设(图3d)。
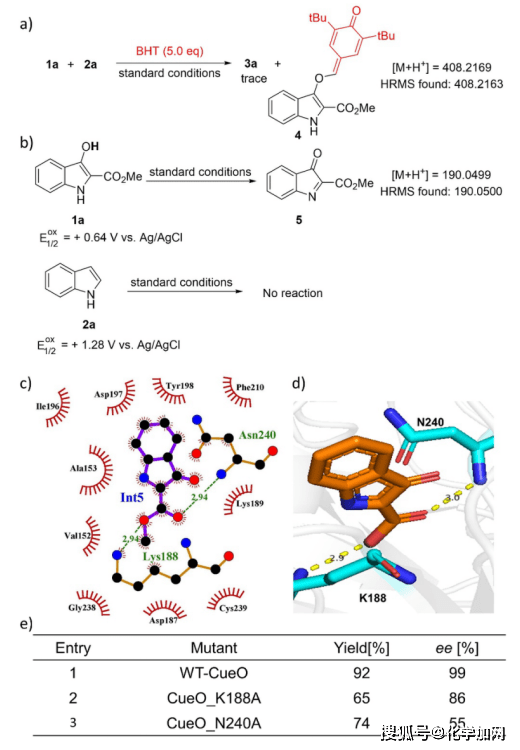
图3 酶促反应机理研究 (来源:Angew. Chem. Int. Ed.)
文献表明,2,2-二取代吲哚啉-3-酮类分子可能具有显著的生物活性。作者利用MTT法测定了部分产物对MCF-7肿瘤细胞系的毒性,得到了两个显著毒性的化合物3f和4a(浓度为50μM)(Figure 4A)。随后的细化实验得到了IC50值分别为34.31 μM和37.2 μM,均高于临床药物伊立替康和5-氟尿嘧啶(Figure 4B)。
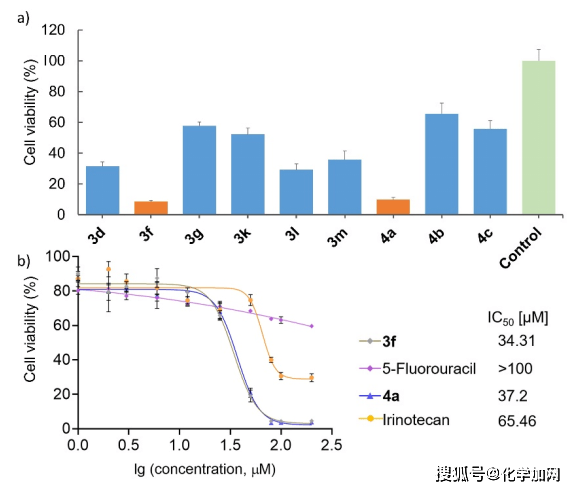
图4 生物活性测试 (来源:Angew. Chem. Int. Ed.)
总结
总之,钟芳锐教授团队以铜酶CueO为生物催化剂,以H2O2为终端氧化剂建立了一种不同吲哚分子间的对映选择性氧化交叉偶联反应,具有操作简便、高原子经济性、较好的底物普适性等特点,提供了一种光学纯2,2-二取代吲哚酮的绿色合成方法,并筛选得到了两个显著抗癌活性分子。该工作拓展了多铜氧化酶的催化杂泛性,将启发铜酶催化其它非天然氧化反应的发展。
Huan Guo, Ningning Sun, Juan Guo, Tai-Ping Zhou, Langyu Tang, Wentao Zhang, Yaming Deng, Rong-Zhen Liao, Yuzhou Wu, Guojiao Wu, Fangrui Zhong*. Angew. Chem. Int. Ed. 2023,doi:10.1002/anie.202219034
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
